Lecture 3: ChemmineR
ChemmineR is a cheminformatics package for analyzing drug-like small molecule data in R. Its latest version contains functions for efficient processing of large numbers of small molecules, physicochemical/structural property predictions, structural similarity searching, classification and clustering of compound libraries with a wide spectrum of algorithms.
## Installation
The R software for running ChemmineR can be downloaded from CRAN (http://cran.at.r-project.org/). The ChemmineR package can be installed from R with:
Loading the Package and Documentation
## Five Minute Tutorial
The following code gives an overview of the most important functionalities provided by ChemmineR. Copy and paste of the commands into the R console will demonstrate their utilities.
Create Instances of SDFset class:
## An instance of "SDFset" with 100 molecules## An instance of "SDFset" with 4 molecules## An instance of "SDF"
##
## <<header>>
## Molecule_Name Source
## "650001" " -OEChem-07071010512D"
## Comment Counts_Line
## "" " 61 64 0 0 0 0 0 0 0999 V2000"
##
## <<atomblock>>
## C1 C2 C3 C5 C6 C7 C8 C9 C10 C11 C12 C13 C14 C15 C16
## O_1 7.0468 0.0839 0 0 0 0 0 0 0 0 0 0 0 0 0
## O_2 12.2708 1.0492 0 0 0 0 0 0 0 0 0 0 0 0 0
## ... ... ... ... ... ... ... ... ... ... ... ... ... ... ... ...
## H_60 1.8411 -1.5985 0 0 0 0 0 0 0 0 0 0 0 0 0
## H_61 2.6597 -1.2843 0 0 0 0 0 0 0 0 0 0 0 0 0
##
## <<bondblock>>
## C1 C2 C3 C4 C5 C6 C7
## 1 1 16 2 0 0 0 0
## 2 2 23 1 0 0 0 0
## ... ... ... ... ... ... ... ...
## 63 33 60 1 0 0 0 0
## 64 33 61 1 0 0 0 0
##
## <<datablock>> (33 data items)
## PUBCHEM_COMPOUND_CID PUBCHEM_COMPOUND_CANONICALIZED PUBCHEM_CACTVS_COMPLEXITY
## "650001" "1" "700"
## PUBCHEM_CACTVS_HBOND_ACCEPTOR
## "7" "..." view(sdfset[1:4]) # Returns summarized content of many SDFs, not printed here
as(sdfset[1:4], "list") # Returns complete content of many SDFs, not printed hereAn SDFset is created during the import of an SD file:
Miscellaneous accessor methods for SDFset container:
## Molecule_Name Source
## "650001" " -OEChem-07071010512D"
## Comment Counts_Line
## "" " 61 64 0 0 0 0 0 0 0999 V2000"## C1 C2 C3 C5 C6 C7 C8 C9 C10 C11 C12 C13 C14 C15 C16
## O_1 7.0468 0.0839 0 0 0 0 0 0 0 0 0 0 0 0 0
## O_2 12.2708 1.0492 0 0 0 0 0 0 0 0 0 0 0 0 0
## O_3 12.2708 3.1186 0 0 0 0 0 0 0 0 0 0 0 0 0
## O_4 7.9128 2.5839 0 0 0 0 0 0 0 0 0 0 0 0 0## C1 C2 C3 C4 C5 C6 C7
## 1 1 16 2 0 0 0 0
## 2 2 23 1 0 0 0 0
## 3 2 27 1 0 0 0 0
## 4 3 25 1 0 0 0 0## PUBCHEM_COMPOUND_CID PUBCHEM_COMPOUND_CANONICALIZED PUBCHEM_CACTVS_COMPLEXITY
## "650001" "1" "700"
## PUBCHEM_CACTVS_HBOND_ACCEPTOR
## "7"Assigning compound IDs and keeping them unique:
## [1] "CMP1" "CMP2" "CMP3" "CMP4"## [1] "650001" "650002" "650003" "650004"## [1] "No duplicates detected!"Converting the data blocks in an SDFset to a matrix:
blockmatrix <- datablock2ma(datablocklist=datablock(sdfset)) # Converts data block to matrix
numchar <- splitNumChar(blockmatrix=blockmatrix) # Splits to numeric and character matrix
numchar[[1]][1:2,1:2] # Slice of numeric matrix## PUBCHEM_COMPOUND_CID PUBCHEM_COMPOUND_CANONICALIZED
## 650001 650001 1
## 650002 650002 1## PUBCHEM_MOLECULAR_FORMULA PUBCHEM_OPENEYE_CAN_SMILES
## 650001 "C23H28N4O6" "CC1=CC(=NO1)NC(=O)CCC(=O)N(CC(=O)NC2CCCC2)C3=CC4=C(C=C3)OCCO4"
## 650002 "C18H23N5O3" "CN1C2=C(C(=O)NC1=O)N(C(=N2)NCCCO)CCCC3=CC=CC=C3"Compute atom frequency matrix, molecular weight and formula:
## MF MW C H N O S F Cl
## 650001 C23H28N4O6 456.4916 23 28 4 6 0 0 0
## 650002 C18H23N5O3 357.4069 18 23 5 3 0 0 0
## 650003 C18H18N4O3S 370.4255 18 18 4 3 1 0 0
## 650004 C21H27N5O5S 461.5346 21 27 5 5 1 0 0Assign matrix data to data block:
## $`650001`
## MF MW C H N O S
## "C23H28N4O6" "456.4916" "23" "28" "4" "6" "0"
## F Cl
## "0" "0"String searching in SDFset:
grepSDFset("650001", sdfset, field="datablock", mode="subset") # Returns summary view of matches. Not printed here.## 1 1 1 1 1 1 1 1 1
## 1 2 3 4 5 6 7 8 9Export SDFset to SD file:
Plot molecule structure of one or many SDFs:
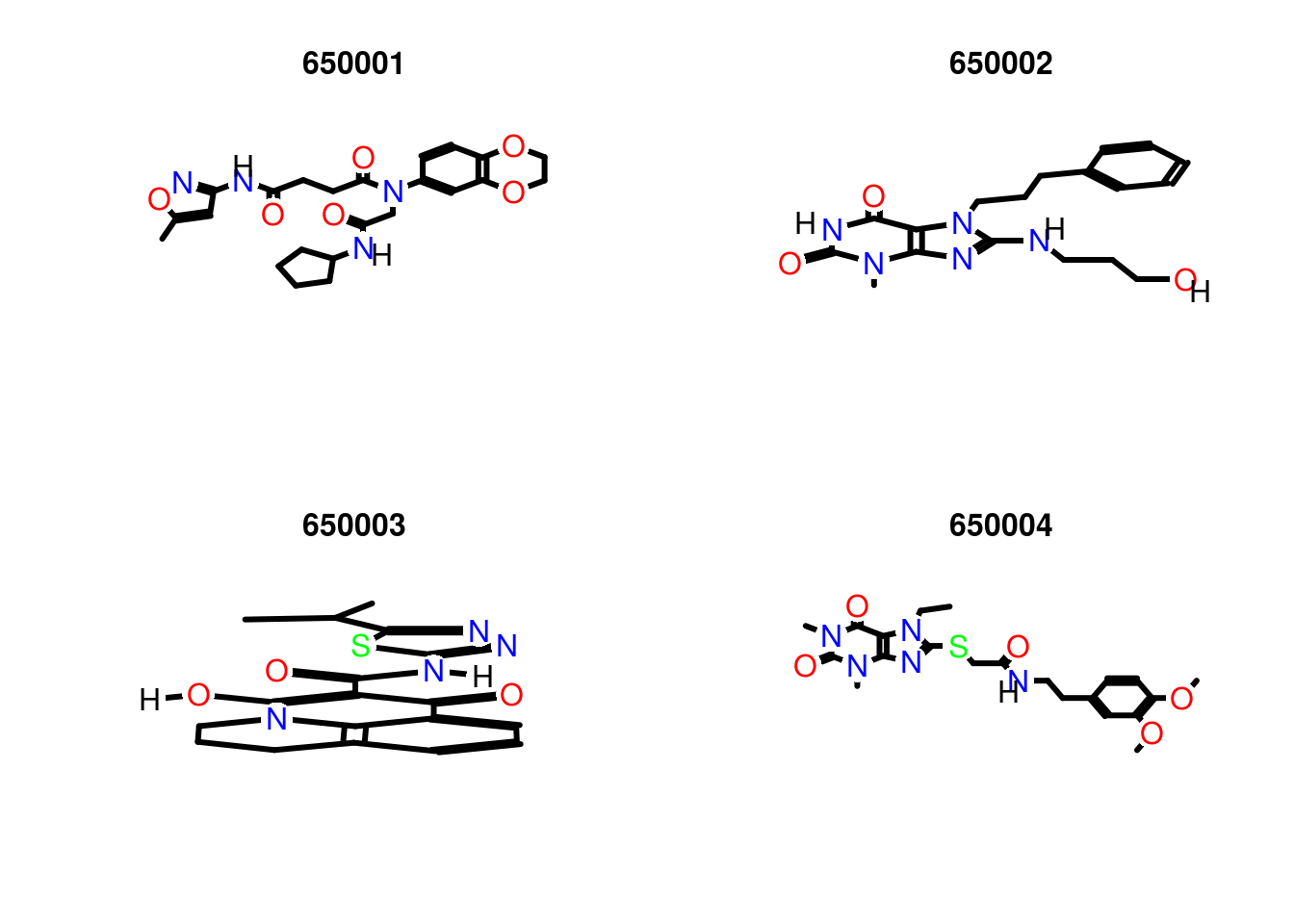

Figure: Visualization webpage created by calling sdf.visualize.
Structure similarity searching and clustering:
data(apset) # Load sample apset data provided by library.
cmp.search(apset, apset[1], type=3, cutoff = 0.3, quiet=TRUE) # Search apset database with single compound.## index cid scores
## 1 1 650001 1.0000000
## 2 96 650102 0.3516643
## 3 67 650072 0.3117569
## 4 88 650094 0.3094629
## 5 15 650015 0.3010753 cmp.cluster(db=apset, cutoff = c(0.65, 0.5), quiet=TRUE)[1:4,] # Binning clustering using variable similarity cutoffs.##
## sorting result...## ids CLSZ_0.65 CLID_0.65 CLSZ_0.5 CLID_0.5
## 48 650049 2 48 2 48
## 49 650050 2 48 2 48
## 54 650059 2 54 2 54
## 55 650060 2 54 2 54### Molecular Structure Data
Classes
SDFstr: intermediate string class to facilitate SD file import; not important for end userSDF: container for single molecule imported from an SD fileSDFset: container for many SDF objects; most important structure container for end userSMI: container for a single SMILES stringSMIset: container for many SMILES strings
Functions/Methods (mainly for SDFset container, SMIset should be coerced with smiles2sd to SDFset)
Accessor methods for
SDF/SDFsetObject slots:
cid,header,atomblock,bondblock,datablock(sdfid,datablocktag)Summary of
SDFset:viewMatrix conversion of data block:
datablock2ma,splitNumCharString search in SDFset:
grepSDFset
Coerce one class to another
- Standard syntax
as(..., "...")works in most cases. For details see R help with?"SDFset-class".
- Standard syntax
Utilities
Atom frequencies:
atomcountMA,atomcountMolecular weight:
MWMolecular formula:
MF…
Compound structure depictions
R graphics device:
plot,plotStrucOnline:
cmp.visualize
Structure Descriptor Data
Classes
AP: container for atom pair descriptors of a single moleculeAPset: container for many AP objects; most important structure descriptor container for end userFP: container for fingerprint of a single moleculeFPset: container for fingerprints of many molecules, most important structure descriptor container for end user
Functions/Methods
Create
AP/APsetinstancesFrom
SDFset:sdf2apFrom SD file:
cmp.parseSummary of
AP/APset:view,db.explain
Accessor methods for AP/APset
- Object slots:
ap,cid
- Object slots:
Coerce one class to another
- Standard syntax
as(..., "...")works in most cases. For details see R help with?"APset-class".
- Standard syntax
Structure Similarity comparisons and Searching
Compute pairwise similarities :
cmp.similarity,fpSimSearch APset database:
cmp.search,fpSim
AP-based Structure Similarity Clustering
Single-linkage binning clustering:
cmp.clusterVisualize clustering result with MDS:
cluster.visualizeSize distribution of clusters:
cluster.sizestat
Folding
- Fold a descriptor with
fold - Query the number of times a descriptor has been folded:
foldCount - Query the number of bits in a descriptor:
numBits
- Fold a descriptor with
Import of Compounds
SDF Import
The following gives an overview of the most important import/export functionalities for small molecules provided by ChemmineR. The given example creates an instance of the SDFset class using as sample data set the first 100 compounds from this PubChem SD file (SDF): Compound_00650001_00675000.sdf.gz (ftp://ftp.ncbi.nih.gov/pubchem/Compound/CURRENT-Full/SDF/).
SDFs can be imported with the read.SDFset function:
data(sdfsample) # Loads the same SDFset provided by the library
sdfset <- sdfsample
valid <- validSDF(sdfset) # Identifies invalid SDFs in SDFset objects
sdfset <- sdfset[valid] # Removes invalid SDFs, if there are anyImport SD file into SDFstr container:
Create SDFset from SDFstr class:
## An instance of "SDFstr" with 100 molecules## An instance of "SDFset" with 100 moleculesSMILES Import
The read.SMIset function imports one or many molecules from a SMILES file and stores them in a SMIset container. The input file is expected to contain one SMILES string per row with tab-separated compound identifiers at the end of each line. The compound identifiers are optional.
Create sample SMILES file and then import it:
data(smisample); smiset <- smisample
write.SMI(smiset[1:4], file="sub.smi")
smiset <- read.SMIset("sub.smi")Inspect content of SMIset:
## An instance of "SMIset" with 100 molecules## $`650001`
## An instance of "SMI"
## [1] "O=C(NC1CCCC1)CN(c1cc2OCCOc2cc1)C(=O)CCC(=O)Nc1noc(c1)C"
##
## $`650002`
## An instance of "SMI"
## [1] "O=c1[nH]c(=O)n(c2nc(n(CCCc3ccccc3)c12)NCCCO)C"Accessor functions:
## [1] "650001" "650002" "650003" "650004"Create SMIset from named character vector:
## An instance of "SMIset" with 2 moleculesExport of Compounds
SDF Export
Write objects of classes SDFset/SDFstr/SDF to SD file:
Writing customized SDFset to file containing ChemmineR signature, IDs from SDFset and no data block:
Example for injecting a custom matrix/data frame into the data block of an SDFset and then writing it to an SD file:
props <- data.frame(MF=MF(sdfset), MW=MW(sdfset), atomcountMA(sdfset))
datablock(sdfset) <- props
write.SDF(sdfset[1:4], file="sub.sdf", sig=TRUE, cid=TRUE)Indirect export via SDFstr object:
sdf2str(sdf=sdfset[[1]], sig=TRUE, cid=TRUE) # Uses default components
sdf2str(sdf=sdfset[[1]], head=letters[1:4], db=NULL) # Uses custom components for header and data blockWrite SDF, SDFset or SDFstr classes to file:
Format Interconversions
The sdf2smiles and smiles2sdf functions provide format interconversion between SMILES strings (Simplified Molecular Input Line Entry Specification) and SDFset containers.
Convert an SDFset container to a SMILES character string:
Convert a SMILES character string to an SDFset container:
When the ChemineOB package is installed these conversions are performed with the OpenBabel Open Source Chemistry Toolbox. Otherwise the functions will fall back to using the ChemMine Tools web service for this operation. The latter will require internet connectivity and is limited to only the first compound given. ChemmineOB provides access to the compound format conversion functions of OpenBabel. Currently, over 160 formats are supported by OpenBabel. The functions convertFormat and convertFormatFile can be used to convert files or strings between any two formats supported by OpenBabel. For example, to convert a SMILES string to an SDF string, one can use the convertFormat function.
This will return the given compound as an SDF formatted string. 2D coordinates are also computed and included in the resulting SDF string.
To convert a file with compounds encoded in one format to another format, the convertFormatFile function can be used instead.
To see the whole list of file formats supported by OpenBabel, one can run from the command-line “obabel -L formats”.
Splitting SD Files
The following write.SDFsplit function allows to split SD Files into any number of smaller SD Files. This can become important when working with very big SD Files. Users should note that this function can output many files, thus one should run it in a dedicated directory!
Create sample SD File with 100 molecules:
Read in sample SD File. Note: reading file into SDFstr is much faster than into SDFset:
Run export on SDFstr object:
write.SDFsplit(x=sdfstr, filetag="myfile", nmol=10) # 'nmol' defines the number of molecules to write to each fileRun export on SDFset object:
Searching PubChem
Get Compounds from PubChem by Id
The function getIds accepts one or more numeric PubChem compound ids and downloads the corresponding compounds from PubChem Power User Gateway (PUG) returning results in an SDFset container. The ChemMine Tools web service is used as an intermediate, to translate queries from plain HTTP POST to a PUG SOAP query.
Fetch 2 compounds from PubChem:
Search a SMILES Query in PubChem
The function searchString accepts one SMILES string (Simplified Molecular Input Line Entry Specification) and performs a >0.95 similarity PubChem fingerprint search, returning the hits in an SDFset container. The ChemMine Tools web service is used as an intermediate, to translate queries from plain HTTP POST to a PubChem Power User Gateway (PUG) query.
Search a SMILES string on PubChem:
Search an SDF Query in PubChem
The function searchSim performs a PubChem similarity search just like searchString, but accepts a query in an SDFset container. If the query contains more than one compound, only the first is searched.
Search an SDFset container on PubChem: